酶[編輯]
維基百科,自由的百科全書
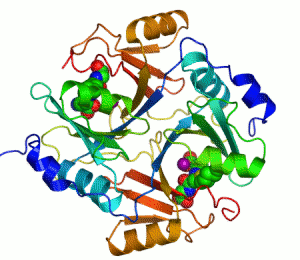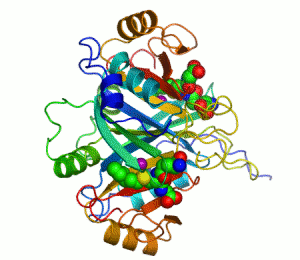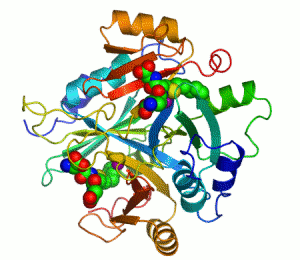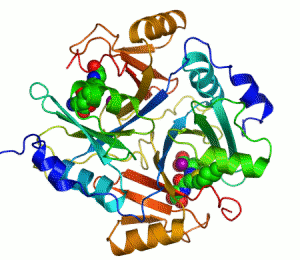
人類乙二醛酶I 的帶狀圖,其催化鋅離子顯示兩個紫色的球體。抑製劑,S-hexylglutathione,則表現為一個空間填充模型,填充兩個活性部位。綠色,紅色,藍色和黃色的球體,分別對應於碳,氧,氮和硫原子。
嘌呤核苷磷酸化酶三維結構的飄帶圖示,不同的胺基酸殘基用不同顏色顯示
酶(德語:Enzym,源於希臘語:ενζυμον,「在酵裡面」;又稱酵素),指具有生物催化功能的高分子物質。[1] 在酶的催化反應體系中,反應物分子被稱為受質,受質通過酶的催化轉化為另一種分子。幾乎所有的細胞活動進程都需要酶的參與,以提高效率。與其他非生物催化劑相似,酶透過降低化學反應的活化能(用Ea或ΔG‡表示)來加快反應速率,大多數的酶可以將其催化的反應之速率提高上百萬倍;事實上,酶是提供另一條活化能需求較低的途徑,使更多反應粒子能擁有不少於活化能的動能,從而加快反應速率。酶作為催化劑,本身在反應過程中不被消耗,也不影響反應的化學平衡。酶有正催化作用,也有負催化作用,不只是加快反應速率,也有減低反應速率。與其他非生物催化劑不同的是,酶具有高度的專一性,只催化特定的反應或產生特定的構型。目前已知的可以被酶催化的反應有約4000種。[2]
雖然酶大多是蛋白質,但少數具有生物催化功能的分子並非為蛋白質,有一些被稱為核酶的RNA分子[3] 和一些DNA分子[4]同樣具有催化功能。此外,通過人工合成所謂人工酶也具有與酶類似的催化活性。[5] 有人認為酶應定義為具有催化功能的生物大分子,即生物催化劑,則該定義中酶包含具有催化功能的蛋白質和核酶。[6]
酶的催化活性可以受其他分子影響:抑制劑是可以降低酶活性的分子;活化劑則是可以增加酶活性的分子。有許多藥物和毒藥就是酶的抑制劑。酶的活性還可以被溫度、化學環境(如pH值)、受質濃度以及電磁波(如微波[7])等許多因素所影響。
酶在工業和人們的日常生活中的應用也非常廣泛。例如,藥廠用特定的合成酶來合成抗生素;加酶洗衣粉通過分解蛋白質和脂肪來幫助除去衣物上的污漬和油漬。
目錄 [隱藏]
1 發現及研究史
2 生物學功能
3 結構與催化機理
3.1 結構
3.2 專一性
3.2.1 「鎖-鑰」模式(「Lock and key」)
3.2.2 誘導契合模式(Induced fit)
3.2.3 群體移動模式(Population shift)
3.3 機理
3.3.1 過渡態的穩定
3.3.2 動態作用
3.4 異位調節
3.5 失活
4 輔因子與輔酶
4.1 輔因子
4.2 輔酶
5 熱力學
6 動力學
7 抑制作用
7.1 可逆抑制作用
7.1.1 競爭性抑制作用(competitive inhibition)
7.1.2 非競爭性抑制作用(non-competitive inhibition)
7.1.3 反競爭性抑制作用(uncompetitive/anti-competitive inhibition)
7.1.4 複合抑制作用
7.2 不可逆抑制作用
7.3 抑制劑的用途
8 活性控制
9 相關疾病
10 命名規則
11 應用
12 參見
13 參考文獻
14 延伸閱讀
15 外部連結
發現及研究史[編輯]
法國科學家路易·巴斯德
酶的發現來源於人們對發酵機理的逐漸了解。早在18世紀末和19世紀初,人們就認識到食物在胃中被消化,[8] 用植物的提取液可以將澱粉轉化為糖,但對於其對應的機理則並不了解。[9]
到了19世紀中葉,法國科學家路易·巴斯德對蔗糖轉化為酒精的發酵過程進行了研究,認為在酵母細胞中存在一種活力物質,命名為「酵素」(ferment)。他提出發酵是這種活力物質催化的結果,並認為活力物質只存在於生命體中,細胞破裂就會失去發酵作用。[10]
1878年,德國生理學家威廉·屈內首次提出了酶(enzyme)這一概念。隨後,酶被用於專指胃蛋白酶等一類非活體物質,而酵素(ferment)則被用於指由活體細胞產生的催化活性。
德國科學家愛德華·比希納
這種對酶的錯誤認識很快得到糾正。1897年,德國科學家愛德華·比希納開始對不含細胞的酵母提取液進行發酵研究,通過在柏林洪堡大學所做的一系列實驗最終證明發酵過程並不需要完整的活細胞存在。[11] 他將其中能夠發揮發酵作用的酶命名為發酵酶(zymase)。[12] 這一貢獻打開了通向現代酶學與現代生物化學的大門,其本人也因「發現無細胞發酵及相應的生化研究」而獲得了1907年的諾貝爾化學獎。在此之後,酶和酵素兩個概念合二為一,並依據比希納的命名方法,酶的發現者們根據其所催化的反應將它們命名。通常酶的英文名稱是在催化受質或者反應類型的名字最後加上-ase的後綴,而對應中文命名也採用類似方法,即在名字最後加上「酶」。例如,乳糖酶(lactase)是能夠剪切乳糖(lactose)的酶;DNA聚合酶(DNA polymerase)能夠催化DNA聚合反應。
人們在認識到酶是一類不依賴於活體細胞的物質後,下一步工作就是鑒定其生化組成成分。許多早期研究者指出,一些蛋白質與酶的催化活性相關;但包括諾貝爾獎得主里夏德·維爾施泰特在內的部分科學家認為酶不是蛋白質,他們辯稱那些蛋白質只是酶分子的攜帶者,蛋白質本身並不具有催化活性。1926年,美國生物化學家詹姆斯·薩姆納完成了一個決定性的實驗。他首次從刀豆得到尿素酶結晶,並證明了尿素酶的蛋白質本質。其後,薩姆納在1931年在過氧化氫酶的研究中再次證實了酶為蛋白質。約翰·霍華德·諾思羅普和溫德爾·梅雷迪思·斯坦利通過對胃蛋白酶、胰蛋白酶和胰凝乳蛋白酶等消化性蛋白酶的研究,最終確認蛋白質可以是酶。以後陸續發現的兩千餘種酶均證明酶的化學本質是蛋白質。以上三位科學家因此獲得1946年度諾貝爾化學獎。[13]
由於蛋白質可以結晶,通過X射線晶體學就可以對酶的三維結構進行研究。第一個獲得結構解析的酶分子是溶菌酶,一種在眼淚、唾液和蛋清中含量豐富的酶,其功能是溶解細菌外殼。溶菌酶結構由大衛·菲利浦(David Phillips)所領導的研究組解析,並於1965年發表。[14] 這一成果的發表標誌著結構生物學研究的開始,高解析度的酶三維結構使得對於酶在分子水平上的工作機制的了解成為可能。
1980年代,托馬斯·切赫和雪梨·奧爾特曼分別從四膜蟲的rRNA前體的加工研究和細菌的核糖核酸酶P複合物的研究中都發現RNA本身具有自我催化作用,並提出了核酶的概念。這是第一次發現蛋白質以外的具有催化活性的生物分子。 1989年,其二人也因此獲得諾貝爾化學獎。[15]
生物學功能[編輯]
在生物體內,酶發揮著非常廣泛的功能。訊息傳遞和細胞活動的調控都離不開酶,特別是激酶和磷酸酶的參與。[16] 酶也能產生運動,通過催化肌球蛋白上ATP的水解產生肌肉收縮,並且能夠作為細胞骨架的一部分參與運送胞內物質。[17] 一些位於細胞膜上的ATP酶作為離子泵參與主動運輸。一些生物體中比較奇特的功能也有酶的參與,例如螢光素酶可以為螢火蟲發光。[18]病毒中也含有酶,或參與侵染細胞(如HIV整合酶和逆轉錄酶),或參與病毒顆粒從宿主細胞的釋放(如流感病毒的神經胺酸酶)。
酶的一個非常重要的功能是參與在動物消化系統的工作。以澱粉酶和蛋白酶為代表的一些酶可以將進入消化道的大分子(澱粉和蛋白質)降解為小分子,以便於腸道吸收。澱粉不能被腸道直接吸收,而酶可以將澱粉水解為麥芽糖或更進一步水解為葡萄糖等腸道可以吸收的小分子。不同的酶分解不同的食物受質。在草食性反芻動物的消化系統中存在一些可以產生纖維素酶的細菌,纖維素酶可以分解植物細胞壁中的纖維素,從而提供可被吸收的養料。
在代謝途徑中,多個酶以特定的順序發揮功能:前一個酶的產物是後一個酶的受質;每個酶催化反應後,產物被傳遞到另一個酶。有些情況下,不同的酶可以平行地催化同一個反應,從而允許進行更為複雜的調控:比如一個酶可以以較低的活性持續地催化該反應,而另一個酶在被誘導後可以較高的活性進行催化。酶的存在確定了整個代謝按正確的途徑進行;而一旦沒有酶的存在,代謝既不能按所需步驟進行,也無法以足夠的速度完成合成以滿足細胞的需要。實際上如果沒有酶,代謝途徑,如糖解作用,無法獨立進行。例如,葡萄糖可以直接與ATP反應使得其一個或多個碳原子被磷酸化;在沒有酶的催化時,這個反應進行得非常緩慢以致可以忽略;而一旦加入己糖激酶,在6位上的碳原子的磷酸化反應獲得極大加速,雖然其他碳原子的磷酸化反應也在緩慢進行,但在一段時間後檢測可以發現,絕大多數產物為葡萄糖-6-磷酸。於是每個細胞就可以通過這樣一套功能性酶來完成代謝途徑的整個反應網路。
結構與催化機理[編輯]
參見:蛋白質結構及酶促反應
丙糖磷酸異構酶(TIM)三維結構的飄帶圖和半透明的蛋白表面圖顯示。丙糖磷酸異構酶是典型的TIM桶摺疊,圖中用不同顏色來表示該酶中所含有的兩個TIM桶摺疊結構域。
作為蛋白質,不同種酶之間的大小差別非常大,從62個胺基酸殘基的4-草醯巴豆酯互變異構酶(4-oxalocrotonate tautomerase)[19] 到超過2500個殘基的動物脂肪酸合酶[20]。酶的三維結構決定了它們的催化活性和機理。[21] 大多數的酶都要比它們的催化受質大得多,並且酶分子中只有一小部分(3-4個殘基)直接參与催化反應。[22] 這些參與催化殘基加上參與結合受質的殘基共同形成了發生催化反應的區域,這一區域就被稱為「活性中心」或「活性位點」。有許多酶含有能夠結合其催化反應所必需的輔因子的結合區域。此外,還有一些酶能夠結合催化反應的直接或間接產物或者受質;這種結合能夠增加或降低酶活,是一種反饋調節手段。
結構[編輯]
與其他非酶蛋白相似,酶能夠摺疊形成多種三維結構類型。有一部分酶是由多個亞基所組成的複合物酶。除了嗜熱菌中的酶以外,大多數酶在高溫情況下會發生去摺疊,其三維結構和酶活性被破壞;對於不同的酶,這種去摺疊的可逆性也有所不同。
專一性[編輯]
三種酶催化機制模式圖:A. 「鎖-鑰匙」模式;B. 誘導契合模式; C. 群體移動模式。
通常情況下,酶對於其所催化的反應類型和受質種類具有高度的專一性。酶的活性位點和受質,它們的形狀、表面電荷、親疏水性都會影響專一性。酶的催化可以具有很高的立體專一性、區域選擇性和化學選擇性(chemoselectivity)。[23] 具體來說,酶只對具有特定空間結構的某種或某類受質起作用。例如,麥芽糖酶只能使α-葡萄糖苷鍵斷裂而對β-葡萄糖苷鍵無影響。此外,酶具有對受質對映異構體的識別能力,只能於一種對映體作用,而對另一對映體不起作用。例如,胰蛋白酶只能水解由L-胺基酸形成的肽鍵,而不能作用於D-胺基酸形成的肽鍵;酵母中的酶只能對D-構型糖(如D-葡萄糖)發酵,而對L-構型無效。
不同酶之間的專一性差別很大。一些酶能夠參與需要有極高準確度的基因組複製和表現中,這些酶都具有「校對」機制。以DNA聚合酶為例,它能夠先完成催化反應,然後再檢測產物是否正確。[24] 這樣一種帶有校對的合成機制,使得具有高保真度的哺乳動物聚合酶的平均出錯幾率低於一百萬分之一,即完成一百萬個反應,出現產物錯誤的反應不到一個。[25] 在RNA聚合酶[26]、氨醯tRNA合成酶[27] 和核醣體[28] 中也發現了類似的校對機制。而對於另一些參與合成次生代謝產物(secondary metabolite)的酶,它們能夠與相對較廣的不同受質作用。有人認為這種低專一性可能對於新的生物合成途徑的進化十分重要。[29]
為了解釋酶的專一性,研究者提出了多種可能的酶與受質的結合模式(後兩種模式為大多數研究者所傾向):
「鎖-鑰」模式(「Lock and key」)[編輯]
該模式由赫爾曼·埃米爾·費歇爾於1894年提出,基於的理論是酶和受質都有一定的外形,若且唯若兩者之間的外形能夠精確互補時,催化反應才可以發生。[30] 這一模式通常被形象地稱為「鎖-鑰匙」模式。雖然這一模式能夠解釋酶的專一性,但卻無法說明為什麼酶能夠穩定反應的過渡態。
誘導契合模式(Induced fit)[編輯]
誘導契合模式詳解圖
該模式由丹尼爾·科什蘭(Daniel Koshland)通過修改「鎖-鑰」模式,於1958年提出。基於的理論是,既然酶作為蛋白質,其結構是具有一定柔性的,因此活性位點在結合受質的過程中,通過與受質分子之間的相互作用,可以不斷發生微小的形變。[31] 在這一模式中,受質不是簡單地結合到剛性的活性位點上,活性位點上的胺基酸殘基的側鏈可以擺動到正確的位置,使得酶能夠進行催化反應。在結合過程中,活性位點不斷地發生變化,直到受質完全結合,此時活性位點的形狀和帶電情況才會最終確定下來。[32] 在一些情況下,受質在進入活性中心時也是會發生微小形變的,如糖苷酶的催化反應。[33]
群體移動模式(Population shift)[編輯]
這一模式是近年來提出的一種新的酶與受質的結合模式,[34] 試圖解釋在一些酶中所發現的受質結合前後,酶的構象有較大變化,而這是用誘導契合模式無法解釋的。其基於的假設是,酶在溶液中同時存在不同構象,一種構象(構象A)為適合受質結合的構象,而另一種(構象B)則不適合,這兩種構象之間保持著動態平衡。在沒有受質存在的情況下,構象B佔主導地位;當加入受質後,隨著受質不斷與構象A結合,溶液中構象A含量下降,兩種構象之間的平衡被打破,導致構象B不斷地轉化為構象A。
機理[編輯]
酶催化機理多種多樣,殊途同歸的是最終都能夠降低反應的ΔG‡:[35]
創造穩定過渡態的微環境。例如,通過與反應的過渡態分子更高的親和力(與受質分子相比),提高其穩定性;或扭曲受質分子,以使得受質更趨向於轉化為過渡態。
提供不同的反應途徑。例如,暫時性地激活受質,形成酶-受質複合物的中間態。
將反應中不同受質分子結合到一起,並固定其方位至反應能夠正確發生的位置,從而降低反應的「門檻」。如果只考慮反應的焓變(ΔH‡),則此作用會被忽略。有趣的是,這一作用同時也會降低反應基態的穩定性,[36] 因此對於催化的貢獻較小。[37]
過渡態的穩定[編輯]
對比同一反應在不受催化和受酶催化的情況,可以了解酶是如何穩定過渡態的。最有效的穩定方式是電荷相互作用,酶可以為過渡態分子上的電荷提供固定的相反電荷,[38] 而這是在水溶液非催化反應體系中不存在的。
動態作用[編輯]
最近的一些研究揭示了酶內部的動態作用與其催化機制之間的聯繫。[39][40] 酶內部的動態作用可以描述為其內部組成元件(小的如一個胺基酸、一組胺基酸;大的如一段環區域、一個α螺旋或相鄰的β鏈;或者可以是整個結構域)的運動,這種運動可以發生在從飛秒(10−15秒)到秒的不同時間尺度。通過這種動態作用,整個酶分子結構中的胺基酸殘基就都可以對酶催化作用施加影響。[41][42][43][44] 蛋白質動態作用在許多酶中都起到關鍵作用,而是小的快速運動還是大的相對較慢的運動起作用更多是依賴於酶所催化的反應類型。對於動態作用的這些新發現,對於了解異位作用、設計人工酶和開發新藥都有重要意義。
但必須指出的是,這種時間依賴的動態進程不大可能幫助提高酶催化反應的速率,因為這種運動是隨機發生的,並且速率常數取決於到達中間態的幾率(P)(P = exp {ΔG‡/RT})。[45] 而且,降低ΔG‡需要相對較小的運動(與在溶液反應中的相應運動相比)以達到反應物與產物之間的過渡態。因此,這種運動或者說動態作用對於催化反應有何貢獻還不清楚。
異位調節[編輯]
主條目:異位調節
在結合效應子的情況下,異位酶能夠改變自身結構,從而達到調節酶活性的效應。這種調節作用可以是直接的,即效應子結合到異位酶上;也可以是間接的,即效應子通過結合其它能夠與異位酶相互作用的蛋白來發揮調節作用。
失活[編輯]
一般情況下,酶在常溫、常壓和中性水溶液條件下可以正常發揮催化活性。在極端條件下,包括高溫、過高或過低pH條件等,酶會失去催化活性,這被稱為酶的失活。但也有一些酶則偏好在非常條件下發揮催化功能,如嗜熱菌中的酶在高溫條件下反而具有較高活性,嗜酸菌中的酶又偏好低pH條件。
輔因子與輔酶[編輯]
主條目:輔酶和輔因子
輔因子[編輯]
並非所有的酶自身就可以催化反應,有一些酶需要結合一些非蛋白小分子後才可以發揮或提高催化活性。[46] 這些小分子被稱為輔因子,它們既可以是無機分子或離子(如金屬離子、鐵硫簇),也可以是有機化合物(如黃素、血紅素)。有機輔因子通常是輔基,可以與其對應的酶非常牢固地結合。這種牢固結合的輔因子與輔酶(如菸鹼醯胺腺嘌呤二核苷酸)不同的是,在整個催化反應過程中,它們一直結合在酶活性位點上而不脫落。
以含有輔因子的碳酸酐酶為例:其輔因子鋅牢固地結合在活性中心,參與催化反應。[47] 黃素或血紅素等輔因子可以參與催化氧化還原反應,往往結合於催化此類反應的酶中。
需要輔因子結合以進行催化的酶,在不結合輔因子的情況下,被稱為酶元(apoenzyme);而在結合了輔因子後,被稱為全酶(holoenzyme)。大多數全酶中,輔因子都是以非共價連接方式與酶結合;也有一些有機輔因子可以與酶共價結合(如丙酮酸去氫酶中的焦磷酸硫胺素)。
輔酶[編輯]
輔酶菸鹼醯胺腺嘌呤二核苷酸的空間填充式結構模型
輔酶是一類可以將化學基團從一個酶轉移到另一個酶上的有機小分子,與酶較為鬆散地結合,對於特定酶的活性發揮是必要的。[46] 有許多維他命及其衍生物,如核黃素、硫胺素和葉酸,都屬於輔酶。[48] 這些化合物無法由人體合成,必須通過飲食補充。不同的輔酶能夠攜帶的化學基團也不同:菸鹼醯胺腺嘌呤二核苷酸或NADP+攜帶氫離子,輔酶A攜帶乙醯基,葉酸攜帶甲醯基,S-腺苷基蛋胺酸也可攜帶甲醯基。[49]
由於輔酶在酶催化反應中其化學組分發生了變化,因此可以認為輔酶是一種特殊的受質或者稱為「第二受質」。這種所謂的第二受質可以被許多酶所利用。例如,目前已知有約七百種酶可以利用輔酶菸鹼醯胺腺嘌呤二核苷酸進行催化。[50]
在細胞內,反應後的輔酶可以被再生,以維持其胞內濃度在一個穩定的水平上。例如,NADPH可以通過磷酸戊糖途徑和甲硫胺酸腺苷基轉移酶作用下的S-腺苷基蛋胺酸來再生。由於輔酶的再生對於維持酶反應體系的穩定是必要的,因此,輔酶再生系統獲得了大量的實驗室以及工業應用。[51]
熱力學[編輯]
參見:活化能、熱力學平衡及化學平衡
有酶或無酶催化反應體系中反應進程與能量關係圖示。可以看出,當反應沒有酶的催化時,受質通常需要獲得較高的活化能才能到達過渡態,然後才能生成產物;而當反應體系中有酶催化時,通過酶對過渡態的穩定作用,降低了達到過渡態所需能量,從而降低了整個反應所需的能量。
與其他催化劑一樣,酶並不改變反應的平衡常數,而是通過降低反應的活化能來加快反應速率(見右圖)。通常情況下,反應在酶存在或不存在的兩種條件下,其反應方向是相同的,只是前者的反應速度更快一些。但必須指出的是,在酶不存在的情況下,受質可以通過其他不受催化的「自由」反應生成不同的產物,原因是這些不同產物的形成速度更快。
酶可以連接兩個或多個反應,因此可以用一個熱力學上更容易發生的反應去「驅動」另一個熱力學上不容易發生的反應。例如,細胞常常通過ATP被酶水解所產生的能量來驅動其他化學反應。
酶可以同等地催化正向反應和逆向反應,而並不改變反應自身的化學平衡。例如,碳酸酐酶可以催化如下兩個互逆反應,催化哪一種反應則是依賴於反應物濃度。[52]
(組織內;高CO2濃度下)
(肺中;低CO2濃度下)
反應式中「*」表示「碳酸酐酶」
當然,如果反應平衡極大地趨向於某一方向,比如釋放高能量的反應,而逆反應不可能有效的發生,則此時酶實際上只催化熱力學上允許的方向,而不催化其逆反應。
動力學[編輯]
主條目:酶動力學
單一受質的酶催化反應機理:酶(表示為「E」)結合受質(表示為「S」),並通過催化反應生成產物(表示為「P」)。
未解決的化學問題:為什麼一些酶的表現快於擴散動力學?
酶動力學是研究酶結合受質能力和催化反應速率的科學。研究者通過酶反應分析法(enzyme assay)來獲得用於酶動力學分析的反應速率資料。
1902年,維克多·亨利提出了酶動力學的定量理論;[53] 隨後該理論得到他人證實並擴展為米氏方程。[54] 亨利最大貢獻在於其首次提出酶催化反應由兩步組成:首先,受質可逆地結合到酶上,形成酶-受質複合物;然後,酶完成對對應化學反應的催化,並釋放生成的產物(見左圖)。
酶初始反應速率(表示為「V」)與受質濃度(表示為「[S]」)的關係曲線。隨著受質濃度不斷提高,酶的反應速率也趨向於最大反應速率(表示為「Vmax」)。
酶可以在一秒鐘內催化數百萬個反應。例如,乳清酸核苷5'-磷酸脫羧酶所催化的反應在無酶情況下,需要七千八百萬年才能將一半的受質轉化為產物;而同樣的反應過程,如果加入這種脫羧酶,則需要的時間只有25毫秒。[55] 酶催化速率依賴於反應條件和受質濃度。如果反應條件中存在能夠將蛋白解鏈的因素,如高溫、極端的pH和高的鹽濃度,都會破壞酶的活性;而提高反應體系中的受質濃度則會增加酶的活性。在酶濃度固定的情況下,隨著受質濃度的不斷升高,酶催化的反應速率也不斷加快並趨向於最大反應速率(Vmax)(見右圖的飽和曲線)。出現這種現象的原因是,當反應體系中受質的濃度升高,越來越多自由狀態下的酶分子結合受質形成酶-受質複合物;當所有酶分子的活性位點都被受質飽和結合,即所有酶分子形成酶-受質複合物時,催化的反應速率達到最大。當然,Vmax並不是酶唯一的動力學常數,要達到一定反應速率所需的受質濃度也是一個重要的動力學指標。這一動力學指標即米氏常數(Km),指的是達到Vmax值一半的反應速率所需的受質濃度(見右圖)。對於特定的受質,每一種酶都有其特徵Km值,表示受質與酶之間的結合強度(Km值越低,結合越牢固,親和力越高)。另一個重要的動力學指標是kcat,定義為一個酶活性位點在一秒鐘內催化受質的數量,用於表示酶催化特定受質的能力。
酶的催化效率可以用kcat/Km來衡量。這一表示式又被稱為特異性常數,其包含了催化反應中所有步驟的反應常數。由於特異性常數同時反映了酶對受質的親和力和催化能力,因此可以用於比較不同酶對於特定受質的 催化效率或同一種酶對於不同受質的催化效率。特異性常數的理論最大值,又稱為擴散極限,約為108至109 M−1s−1;此時,酶與受質的每一次碰撞都會導致受質被催化,因此產物的生成速率不再為反應速率所主導,而分子的擴散速率起到了決定性作用。酶的這種特性被稱為「催化完美性」或「動力學完美性」。相關的酶的例子有磷酸丙糖異構酶、碳酸酐酶、乙醯膽鹼酯酶、過氧化氫酶、延胡索酸酶、β-內醯胺酶和超氧化物歧化酶。
米氏方程是基於質量作用定律而確立的,而該定律則基於自由擴散和熱動力學驅動的碰撞這些假定。然而,由於酶/受質/產物的高濃度和相分離或者一維/二維分子運動,許多生化或細胞進程明顯偏離質量作用定律的假定。[56] 在這些情況下,可以應用分形米氏方程。[57][58][59][60]
存在一些酶,它們的催化產物動力學速率甚至高於分子擴散速率,這種現象無法用目前公認的理論來解釋。有多種理論模型被提出來解釋這類現象。其中,部分情況可以用酶對受質的附加效應來解釋,即一些酶被認為可以通過雙偶極電場來捕捉受質以及將受質以正確方位擺放到催化活性位點。另一種理論模型引入了基於量子理論的穿隧效應,即質子或電子可以穿過激活能壘(就如同穿過隧道一般),但關於穿隧效應還有較多爭議。[61][62] 有報導發現色胺中質子存在量子穿隧效應。[63] 因此,有研究者相信在酶催化中也存在著穿隧效應,可以直接穿過反應能壘,而不是像傳統理論模型的方式通過降低能壘達到催化效果。有相關的實驗報導提出在一種醇去氫酶的催化反應中存在穿隧效應,[64] 但穿隧效應是否在酶催化反應中普遍存在並未有定論。[65]
抑制作用[編輯]
主條目:酶抑制劑
酶的催化活性可以被多種抑制劑所降低。
不同的抑制類型。分類參考自[66]。圖中,「E」表示酶;「I」表示抑制劑;「S」表示受質;「P」表示產物。
可逆抑制作用[編輯]
可逆抑制作用的類型有多種,它們的共同特點在於抑制劑對酶活性的抑制反應具有可逆性。
競爭性抑制作用(competitive inhibition)[編輯]
抑制劑與受質競爭結合酶的活性位點(抑制劑和受質不能同時結合到活性位點)。對於競爭性抑制作用,催化反應的最大反應速率值沒有變,但是需要更高的受質濃度,反映在表觀Km值的增加。
非競爭性抑制作用(non-competitive inhibition)[編輯]
非競爭性抑制抑制劑可以與受質同時結合到酶上,即抑制劑不結合到活性位點。酶-抑制劑複合物(EI)或酶-抑制劑-受質複合物(EIS)都沒有催化活性。與競爭性抑制作用相比,非競爭性抑制作用不能通過提高受質濃度來達到所需反應速度,即表觀最大反應速率Vmax的值變小;而同時,由於抑制劑不影響受質與酶的結合,因此Km值保持不變。
反競爭性抑制作用(uncompetitive/anti-competitive inhibition)[編輯]
反競爭性抑制作用比較少見:抑制劑不能與處於自由狀態下的酶結合,而只能和酶-受質複合物(ES)結合,在酶反應動力學上表現為Vmax和Km值都變小。這種抑制作用可能發生在多亞基酶中。
複合抑制作用[編輯]
這種抑制作用與非競爭性抑制作用比較相似,區別在於EIS複合物殘留有部分酶的活性。在許多生物體中,這類抑制劑可以作為負反饋機制的組成部分。若一個酶體系生產了過多的產物,那麼產物就會抑制合成該產物的酶體系中第一個酶的活性,這就可以保證一旦合成足夠多的產物後,該產物的合成速率會下降或停止。受這種抑制作用調控的酶通常為多亞基酶,並具有與調控產物結合的異位結合位點。這種抑制作用的反應速率與受質濃度的關係圖不再是雙曲線形而是S形。
不可逆抑制作用[編輯]
不可逆抑制劑可以與酶結合形成共價連接,而其他抑制作用中酶與抑制劑之間都是非共價結合。這種抑制作用是不可逆的,酶一旦被抑制後就無法再恢復活性狀態。這類抑制劑包括二氟甲基鳥胺酸(一種可用於治療寄生蟲導致的昏睡症的藥物[67])、苯甲基磺醯氟(PMSF)、青霉素和阿司匹林。這些藥物都是與酶活性位點結合併被激活,然後與活性位點處的一個或多個胺基酸殘基發生不可逆的反應形成共價連接。
抑制劑的用途[編輯]
酶抑制劑常被用作藥物,同樣也可以被作為毒藥使用。而藥物和毒藥之間的差別通常非常小,大多數的藥物都有一定程度的毒性,正如帕拉塞爾蘇斯所言:「所有東西都有毒,沒有什麼是無毒的」(「In all things there is a poison, and there is nothing without a poison」)。[68] 相同的,抗生素和其他抗感染藥物只是特異性地對病原體而不是對宿主有毒性。
一個獲得廣泛應用的抑制劑藥物是阿司匹林,它可以抑制環加氧酶的活性,而環加氧酶可以生產炎症反應信使前列腺素,因此,阿司匹林可以起到抑制疼痛與炎症的作用。而劇毒毒藥氰化物可以通過結合細胞色素氧化酶位點處的銅和鐵原子不可逆地抑制酶活性,從而抑制細胞的呼吸作用。[69]
活性控制[編輯]
細胞內有五種控制酶催化活性的機制:
根據外界環境的變化,細胞可以增強或減弱酶的生產(即酶相關基因的轉錄和轉譯)。這屬於一種基因調控,被稱為酶的誘導和抑制。例如,當環境中出現如青霉素這樣的抗生素時,部分細菌可以對抗生素產生抗性,其原因就在於細菌體內的β-半乳糖苷酶被誘導而大量生產,這種酶可以水解青霉素分子上關鍵的β-乳胺環。另一個例子是在人體肝臟中存在一類酶對於藥物代謝非常重要的酶,細胞色素P450氧化酶;對這一類酶的誘導或抑制,會導致藥物相互作用。
通過將特定的酶分隔在特定的細胞組分中,細胞可以完成不同的代謝途徑。例如,脂肪酸的合成是由細胞溶質、內質網和高基氏體中的一系列酶所完成,而脂肪酸的降解(以提供能量)是在粒線體中由另一系列酶通過β-氧化來完成。[70]
酶可以被抑制劑與活化劑所調控。例如,一個代謝途徑中的終產物常常是這一途徑中第一個酶的抑制劑,從而調控這一代謝途徑的產物量。這種調控機制被稱為負反饋機制,因為終產物的合成量是受其自身濃度調控。負反饋機制可以根據細胞的需要,有效地調節中間代謝物的合成速率,從而使細胞的能量和物質的分配更為高效,並防止多餘產物的合成。控制酶的作用,可以在生物體內維持一個穩定的內部環境(即體內平衡)。
轉譯後修飾也可以調控酶的活性。這些修飾包括磷酸化、肉豆蔻酸化和糖基化。例如,細胞接受胰島素信號後,對包括糖原合酶在內的多個酶進行磷酸化,幫助控制糖原的合成或降解,使得細胞可以對血糖的變化產生反應。[71] 另一個轉譯後修飾的例子是多肽鏈的剪切。胰凝乳蛋白酶,一種消化性蛋白酶,是產生於胰臟中的無活性的胰凝乳蛋白酶原,這一蛋白通過運輸到達胃後才被激活。這種方式有效地防止了胰凝乳蛋白酶在進入腸之前消化胰臟或其他組織。這種無活性的酶的前體被命名為酶原。
還有一些酶可以通過定位到不同環境後而被激活,比如從還原態的環境(細胞質)到氧化態環境(細胞周質空間),從高pH環境到低pH環境等。流感病毒的紅血球凝集素蛋白就是一個例子:當它接觸到宿主細胞囊泡的酸性環境時,它的構象立刻發生變化,導致其獲得激活。[72]
相關疾病[編輯]
酶的活性必須嚴格控制以維持體內平衡,對於能夠影響一個關鍵酶的功能的任何基因缺陷(如突變導致活性變化,過量表現、過低表現或刪除突變)都可能導致遺傳性疾病發生。許多事實顯示,一種致命疾病的病因可以只是由於人體中的數千種酶中的一種發生功能故障。
苯丙酮尿症:此種病症是典型的酶相關病例之一。病因是苯丙胺酸羥化酶(其功能是催化苯丙胺酸降解過程中的第一步)上一個胺基酸位點發生了突變,導致體內苯丙胺酸和相關產物的水平過高,如果沒有得到合適的治療,會進一步導致智能障礙。
紫質病:該病是由於血紅素生物合成途徑中特定酶的酶活性過低(基因突變或其他原因導致),使得中間產物紫質的產生和排泄異常,在一定誘因(如陽光照射)下,可導致皮膚或其他組織器官發生病變。
當生殖細胞中編碼DNA修復相關酶的基因發生突變,其結果會導致遺傳性癌症綜合病徵,如著色性干皮症。DNA修復酶的缺陷導致人體喪失修復突變基因的能力。發生的突變不斷積累,最終使得患者有多種癌症發生。
命名規則[編輯]
參見:EC編號
酶通常是根據其受質性質或其所催化的化學反應類型來命名(英文中,在單詞的最後要加上-ase的後綴),如乳糖酶(lactase)、醇去氫酶(alcohol dehydrogenase)和DNA聚合酶(DNA polymerase)。但這樣往往會導致具有相同功能的不同的酶(同工酶)有相同的名字,而同工酶有著不同的胺基酸序列並可以通過它們不同的最適pH和不同的反應動力學參數來區分。而且,一些酶具有兩個或多個類型催化活性,這種命名方式就會使得同一種酶具有不同的名字。為了解決這些問題,國際生物化學與分子生物學聯盟發展出了酶的系統命名法:
EC編號規定每種酶都由四個數字來表示,並在數字前冠以「EC」。其中,第一個數字是根據酶促反應的性質來將酶大致分為六大類:
EC 1,氧化還原酶:催化受質進行氧化還原反應的酶類。例如,乳酸去氫酶、琥珀酸去氫酶、細胞色素氧化酶、過氧化氫酶、過氧化物酶等。
EC 2,轉移酶:催化受質之間進行某些基團的轉移或交換的酶類。例如,甲基轉移酶、氨基轉移酶、己糖激酶、磷酸化酶等。
EC 3,水解酶:催化受質發生水解反應的酶類。例如,澱粉酶、蛋白酶、脂肪酶、磷酸酶等。
EC 4,解離酶:催化從受質中移去一個基團並留下雙鍵的反應或其逆反應的酶類。例如,碳酸酐酶、醛縮酶、檸檬酸合酶等。
EC 5,異構酶:催化各種同分異構體之間的相互轉化的酶類。例如,磷酸丙糖異構酶、消旋酶等。
EC 6,連接酶:催化兩分子受質合成為一分子化合物,同時偶聯有ATP的磷酸鍵斷裂釋放能量的酶類。例如,谷氨醯胺合成酶、胺基酸:tRNA連接酶等。
國際系統分類法除按上述六類將酶依次編號外,還根據酶所催化的化學鍵的特點和參加反應的基團的不同,將每一大類又進一步分類。編號中的第一個數字表示該酶屬於六大類中的哪一類;第二個數字表示該酶屬於哪一亞類;第三個數字表示亞-亞類;第四個數字是該酶在亞-亞類中的排序。
一些酶的命名舉例
編號 推薦命名 系統命名 催化的反應
EC 1.4.1.3 谷胺酸去氫酶 L-谷胺酸:NAD+氧化還原酶 L-谷胺酸+H2O+NAD+⇔α-酮戊二酸+NH3+NADH
EC 2.6.1.1 天門冬胺酸氨基轉移酶 L-天門冬胺酸:α-酮戊二酸氨基轉移酶 L-天門冬胺酸+α-酮戊二酸⇔草醯乙酸+L-谷胺酸
EC 3.5.3.1 精胺酸酶 L-精胺酸脒基水解酶 L-精胺酸+H2O⇒L-鳥胺酸+尿素
EC 4.1.2.13 果糖二磷酸醛縮酶 D-果糖-1,6-二磷酸:D-甘油醛-3-磷酸解離酶 D-果糖-1,6-二磷酸⇔磷酸二羥丙酮+D-甘油醛-3-磷酸
EC 5.3.1.9 磷酸葡萄糖異構酶 D-葡萄糖-6-磷酸酮醇異構酶 D-葡萄糖-6-磷酸⇔D-果糖-6-磷酸
EC 6.3.1.2 谷氨醯胺合成酶 L-谷胺酸:氨連接酶 ATP+L-谷胺酸+NH3⇒ADP+磷酸+L-谷氨醯胺
應用[編輯]
酶被用於化工等各類需要高度特異性催化情況的用途。但是,酶通常能夠催化的反應數量有限,而且它們在無機溶液中和高溫情況下缺乏穩定性。為了提高酶的應用性,利用蛋白質工程通過合理設計或體外進化來造出具有新特點(例如耐高溫)的酶已經成為一個活躍的研究領域。[73][74] 這類研究工作也有了成功的例子,一些能夠催化自然界中的酶所無法催化的反應的酶已經開始被設計出來。[75]
應用 所用酶 用途
烹飪業
α-澱粉酶催化澱粉分解為蔗糖
真菌(一般為酵母)α-澱粉酶(在烘烤過程中可以被破壞)[來源請求] 催化麵粉中的澱粉分解為蔗糖。在這一過程中,酵母會產生二氧化碳。可用於饅頭、麵包以及其他一些中西式糕點的製作。
蛋白酶 製作餅乾的過程中,通常用它來降低麵粉中蛋白質的含量。
木瓜蛋白酶 將肉嫩化,以利於烹飪。
嬰兒食品 胰蛋白酶 經過酶處理的嬰兒食品更易於消化。
釀酒業
麥芽
麥芽中的酶 將澱粉和蛋白質降解為糖、胺基酸和肽段,而這些原料可以被酵母發酵產生酒精。
工業生產的麥酶 廣泛用於替代天然酶進行啤酒釀造
澱粉酶、葡聚糖酶和蛋白酶 分解麥芽中的多聚糖鏈和蛋白質。
β-葡聚糖酶和阿拉伯木聚糖酶 提高麥汁和啤酒的濾過性。
澱粉葡糖苷酶和普魯蘭酶 製造低卡路里啤酒和調整發酵能力。
蛋白酶 除去在啤酒儲存過程中產生的絮狀物。
乙醯乳酸脫羧酶(ALDC) 避免丁二酮的產生。
果汁製造業 纖維素酶、果膠酶 降解果汁中的不溶物。
乳品業
洛克福羊乳酪
凝乳酶,提自反芻動物(牛、羊)幼崽的胃。 製造乳酪,水解蛋白質。
用微生物生產的凝乳酶 越來越多地使用於乳品製造業。
脂酶 洛克福羊乳酪的製作過程中添加,以加快乳酪的成熟。
乳糖酶 將乳糖分解為葡萄糖和半乳糖。
澱粉加工業 澱粉酶、澱粉葡糖苷酶和葡糖糖化酶 將澱粉轉化為葡萄糖和各種糖漿。
葡萄糖異構酶 在生產高果糖含量的糖漿時,將葡萄糖轉化為果糖。這樣生產的糖漿有更好的甜度和更低的卡路里含量(與同樣甜度的蔗糖相比)。
造紙業
造紙廠
澱粉酶、木聚糖酶、纖維素酶和木質酶 澱粉酶用於降解澱粉至低粘性,加膠和給紙加膜;木聚糖酶能夠降低脫色過程所需的漂白劑;纖維素酶使纖維光滑並增強紙的排水性;木質酶可以消除木質素以軟化紙質。
生物燃料生產
纖維素的三維結構
纖維素酶 用於降解纖維素,產生可用於發酵的蔗糖。(參見en:cellulosic ethanol)。
木質酶 用於木質廢品的降解。
生物去垢劑 主要為蛋白酶(產自細菌的膜外部分) 用於洗衣過程中的浸泡階段,幫助除去衣物上的含有蛋白質的污漬。
澱粉酶 除去洗衣機上的澱粉殘餘。
脂酶 幫助除去衣物上的油漬。
纖維素酶 作為生物纖維(如棉質衣物)的柔順劑。
隱形眼鏡清潔劑 蛋白酶 清除隱形眼鏡上的蛋白質,以防止細菌滋生。
橡膠製造業 過氧化氫酶 催化過氧化物產生氧氣,以將膠乳轉化為泡沫橡皮。
攝影業 蛋白酶(無花果蛋白酶) 溶解底片上的明膠,使得銀成分顯現。
分子生物學
DNA雙螺旋中的一部分
限制內切酶、DNA連接酶和聚合酶 用於在遺傳工程進行基因操縱,對於藥理學、農業和醫學有重要意義。在限制性酶切和聚合酶鏈鎖反應上獲得廣泛應用。分子生物學方法在法醫學的鑒定實驗上也有重要應用。